I develop methods to analyze genome sequencing data in the context of other ‘omics and clinical health data to prioritize and functionally interpret genetic variants with roles in human disease. See my CV for details.
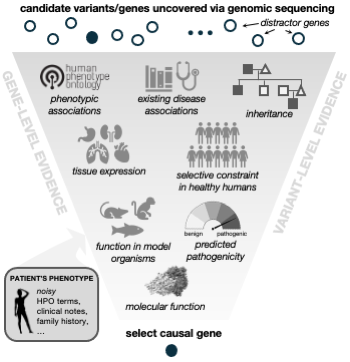 Image from our Nat Comm, 2023 paper.
Image from our Nat Comm, 2023 paper.
Functional genomics for case-based, “N-of-1” analyses
The genome is a big space, and accurately pinpointing variants that underlie specific human health conditions is a formidable challenge. Traditionally, genes have been treated as black box functional units, but we now know that individual variants within and between genes can have wildly different impacts on phenotypes and disease. Because comprehensive, in vivo (in a living system) functional assessment of all possible genetic variants is often infeasible, we instead turn to in silico (computational) variant functionality predictions. We develop integrative tools for assessing the functionality of specific genomic positions and are interested in leveraging multimodal biological and biomedical data to derive new insights on the functional impact of genetic variants. [30535108, 33580225]
Integration of clinical phenotyping
Patient clinical phenotyping data is an essential component in interpreting the impact of genetic variants on human health. Phenotyping data can be noisy, unstructured, and difficult to obtain, and utilizing this information often requires deep clinical intuition. We are interested in developing computational approaches for streamlining the extraction and curation of standardized phenotype data, and leveraging this data for improving diagnostic gene prioritization and interpretation. [37828001, 40542121, 40770127]
Deriving insights from population-level analyses
Even though the genome is a big space, it is also a finite space with respect to simple (single nucleotide and short insertion/deletion) variants, which are the most easily detected and interpreted variant class. This means that as the number of sequenced genomes continues to grow, we will begin to see a saturation of all possible simple variants that are compatible with life, as well as recurrence of disease-relevant variants in phenotypically-matched patient cohorts. Indeed, the number of sequenced tumor genomes has surpassed 10s of thousands; collective cohorts of sequenced Mendelian patients is exceeding 100s of thousands; and ancestrally-diverse, control cohorts of healthy sequenced individuals is set to pass a million. By integrating predicted and experimentally-derived variant functionality information, evolutionary signals of selection and constraint, and accurate mutational models, we will have the power to detect extremely rare variants that play roles in human cancers and other diseases by jointly analyzing these sequenced cohorts. [32711844, 40770127]
Publications
![]() = project lead,
= project lead, ![]() = corresponding author,
= corresponding author, ![]() = team science
= team science
- Joint, multifaceted genomic analysis enables diagnosis of diverse, ultra-rare monogenic presentations. Nature Communications, 2025.

- Simulation of undiagnosed patients with novel genetic conditions. Nature Communications, 2023.

- Commonalities across computational workflows for uncovering explanatory variants in undiagnosed cases. Genetics in Medicine, 2021.
- Innovative methodological approaches for data integration to derive patterns across diverse, large-scale biomedical datasets. Pac Symp Biocomput, 2021.
- How medical mysteries push back the frontier of genomics knowledge. UDN PEER Newsletter, 2021.
- PertInInt: An integrative, analytical approach to rapidly uncover cancer driver genes with perturbed interactions and functionalities. Cell Systems, 2020.
- Ongoing challenges and innovative approaches for recognizing patterns across large-scale, integrative biomedical datasets. Pac Symp Biocomput, 2020.
- Systematic, domain-based aggregation of protein structures highlights DNA-, RNA-, and other ligand-binding positions. Nucleic Acids Research, 2019.
- Pervasive variation of transcription factor orthologs contributes to regulatory network divergence. PLoS Genetics, 2015.
- Formatt: Correcting protein structural alignments by sequence peeking. ACM-BCB’11, 2011.
- Enriched phenotypes in rare variant carriers suggest pathogenic mechanisms in rare disease patients. BioData Mining, 2025.
- VarPPUD: Variant post prioritization developed for undiagnosed genetic disorders. PLoS Comput Biol, 2025.
- Few-shot learning for phenotype-driven diagnosis of patients with rare genetic diseases. npj Digital Medicine, 2025.

- An optimized variant prioritization process for rare disease diagnostics: recommendations for Exomiser and Genomiser. Genome Biology, 2025.
- Phenotypic prediction of missense variants via deep contrastive learning. Nature Biomedical Engineering, 2025.
- A genome-wide approach for the discovery of novel repeat expansion disorders in the Undiagnosed Diseases Network cohort. Genetics in Medicine, 2025.
- RExPRT: a machine learning tool to predict pathogenicity of tandem repeat loci. Genome Biology, 2024.
- Polygenic risk scores for autoimmune related diseases are significantly different and skewed in cancer exceptional responders. npj Precision Oncology, 2024.
- The contribution of mosaicism to genetic diseases and de novo pathogenic variants. Am J Med Genet Part A, 2023.
- Formatt: Correcting protein structural alignments by incorporating sequence alignment. BMC Bioinformatics, 2012.
- Evolving soft robotic locomotion in PhysX. ACM-GECCO’09, 2009.